
核磁気共鳴(NMR)とは,強い磁場中に測定試料をおき,核スピンの向きをそろえた分子にラジオ波を照射することで得られるスペクトル情報から,分子構造などなどを解析する分析手法です。
合成系であれば,毎日といってよいぐらい測定する装置で,私も簡単な分子ではあるものの合成していたので,NMRにはさんざんお世話になった。昔は「シューーーースポンッ!」と自動サンプラーで測定を開始してくれるアタッチメントがなかったので,夜中にNMR室に行って交換したものである。。。
今回は,研究時間を有効に活用するための時短テクニックと言える。以前の記事で,スペクトルデータサイトのを紹介した記事はコチラである。合わせて読んでもらいたい。>>SDBS&NIST等を用いた化学スペクトルの見つけ方・使い方
まず初めに
さて,今回は構造がわかっている分子の1H-NMR,13C-NMR,COSY,HMBC/HSCQを予想できるフリーソフトの紹介とその使い方である。
▼ 無償のNMRスペクトル予測サービスのnmrdb.org
コチラをクリック→→ Simulate and predict NMR spectra (nmrdb.org)
▼

「これで,あの解析の苦労がなくなる・・」ではないが,毎日のようにNMRを使わない研究者にとっては,
・オルト位とパラ位ってどっちが先だっけ?
・ここに電子供与性の置換基がついたから・・・
・OHのHって検出されるんだっけ???
の様に,
学部の機器分析を思い出して,正解かどうかわからないことに時間を割くよりは「サクッ」とニューラルネットワークの力を借りて,「正解」あるいは「正解に近い」スペクトル結果を予想した後,それを鵜呑みにはしないつつも参考にしながら解析に利用した方が研究時間を有効に利用できるといったものである。
NMRの授業動画
▼ とは言いつつも,ちゃんとNMRの原理をわかったうえで,予想された結果を見た方が良い・早いに決まっているのである。NMRの原理については,下記の授業動画を紹介しておく。学部の授業であれば十二分の内容である。
こういったソフトの性能を思う存分引き出すには普段の学びが重要
なお,NMRの結果予想で有名なソフトと言えば”ChemDraw”であろう。高いんだけどね!学生の時にはChemDrawのNMR予想も大変お世話になったものです。>> 名称を変更してChemBioDrawですね
当ラボは,ChemSketchを利用しています。ACD/ChemSketch Freeware 12.0は個人での利用が無償で行える化学構造式描画ソフトです。>> ダウンロードはコチラ →→ ACD/Lab ChemSketch
nmrdb.orgについて
予想フリーソフトnmrdb.orgと注意点
構造がわかっている分子構造のNMRを予想してくれます。いい加減な予想ではなくニューラルネットワークを用いたアルゴリズムで予想してくれます。ChemDraw同様に外れることもありますが,高濃度や共存物質の影響が大きいなどの変な条件でなければシフト位置が多少ズレるくらいなので,解析の第一歩にはもってこいです。
ただし,1点だけ注意点があります!
nmrdb.orgはWikipedaのようなオープンデータベースを目指しており、予想した構造はSPINUSという別のウェブサービスにも自動で送信されます。そのため,特許等に関連する新規の分子構造を検索することは控えることをお勧めします。
指導教員と相談して使用してくださいね!
※ 当ラボの場合は,市販の試薬あるいはちょっとした変更なので,使っても構いません。
nmrdb.orgの使い方
登録不要です。ウエブサイト上で完結できるフリーソフトです。なお,しつこいですが検索した分子構造はオープンサイトであるSPINUSに送信されるので,特許等に係る場合は気を付けてくださいね。
1.アクセス
次のURLにアクセスしてください。 >> https://www.nmrdb.org/
2.メニューバーから選択
上部のメニューバー”About”, ”Predict 1H NMR”, ”Predict 2D”,”Tools”,”Exercies”から予想したいスペクトルの種類を選んで選択する。
今回は 1H-NMRを使いたいので,”Predict 1H NMR”をクリックする。
5-10秒ほど待つ!
※初めて使うときは データを転送するけど良いか?みたいなウィンドウが出るので「I agree」を押す。
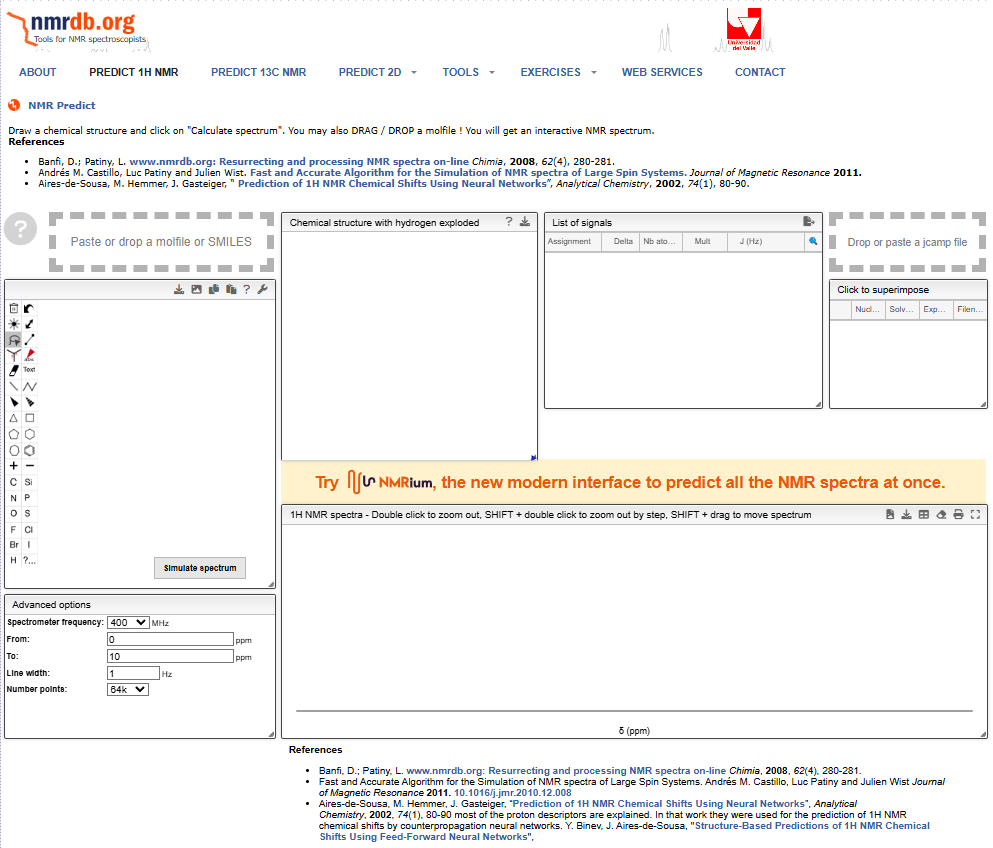
予測するためのウィンドウが8個ほど出てきます。
3.分子構造の入力(作成)
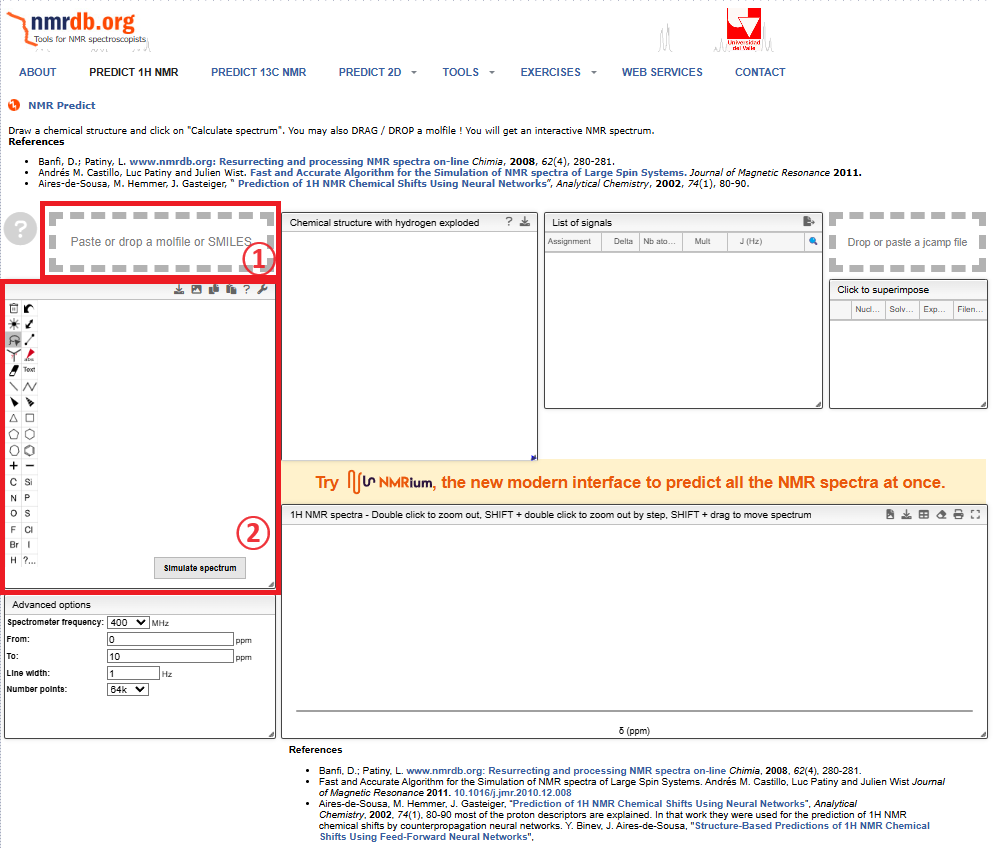
左端の②のウィンドウ内で分子構造を作成できるので,頑張って描く!
or
画面が小さくて目がしばしばするので,
私はChemSketchで描いて,”mol”ファイルで保存して,①にドロップすることで分子構造を読み込んでくれます。
①にckemsketchで書いた分子構造をドロップするか。
②で分子構造を書くか。
4. 入力後に予想ボタンを押す
chemsketchで分子構造を作成して”mol”ファイルで保存してドロップしたのが以下の画像です。
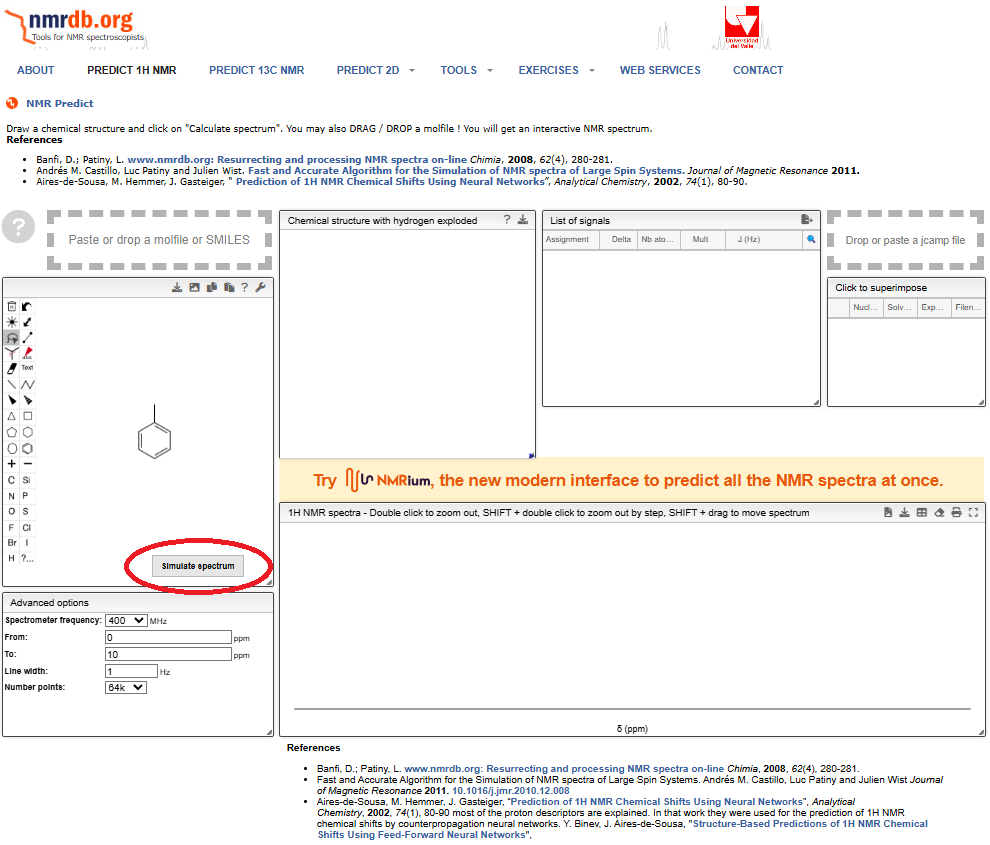
「ペロッ」と入るので,右下の”simulate spectrum”を押すと予想結果が出てきます。
数秒待つ!
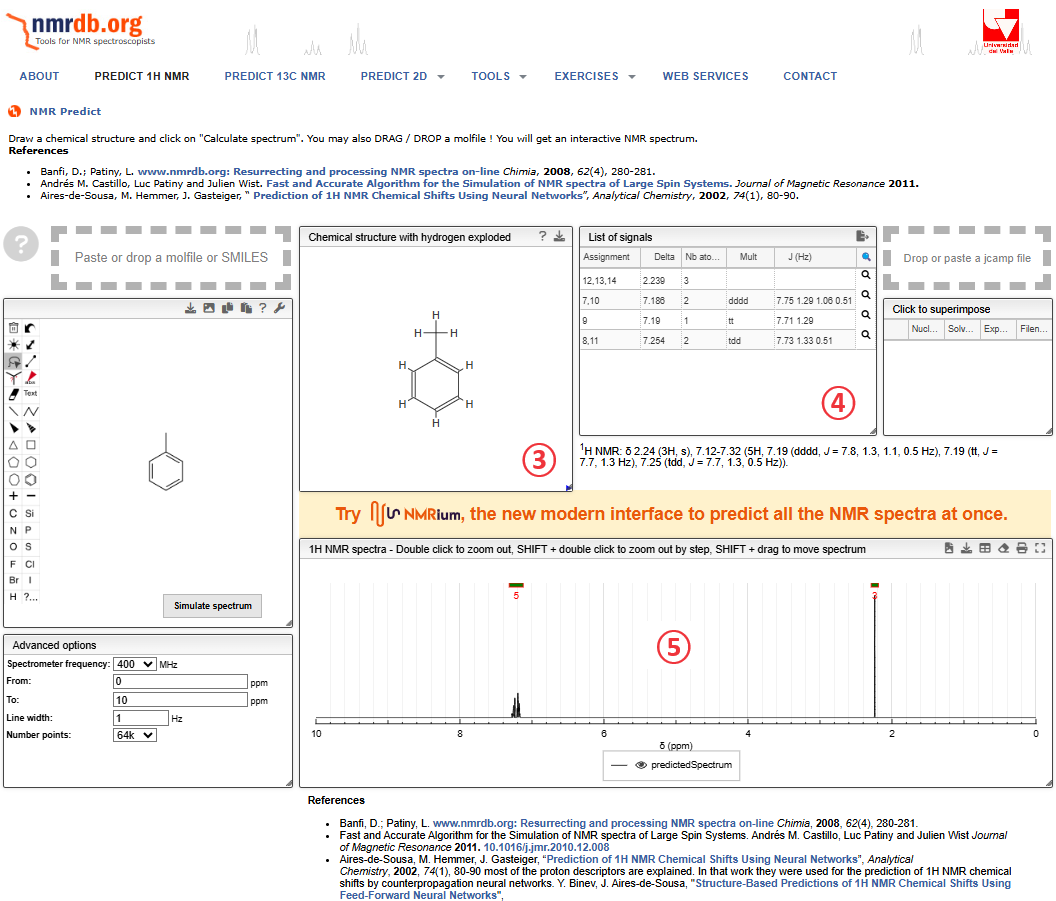
先ほどまで空欄だったウィンドウに
③ 省略されたプロトンが記載された分子構造の表記
④ 予想されたプロトンの化学シフト(ppm)などのデータが記載
⑤ 実際の予想されたNMRチャートが表記
5.予想結果の見方
予想されたらデータの見方であるが,マウスの先端を③のプロトンに合わせるか④の行に合わせると,③~⑤が連動して”ハイライト(黄色い蛍光)”が出ます。
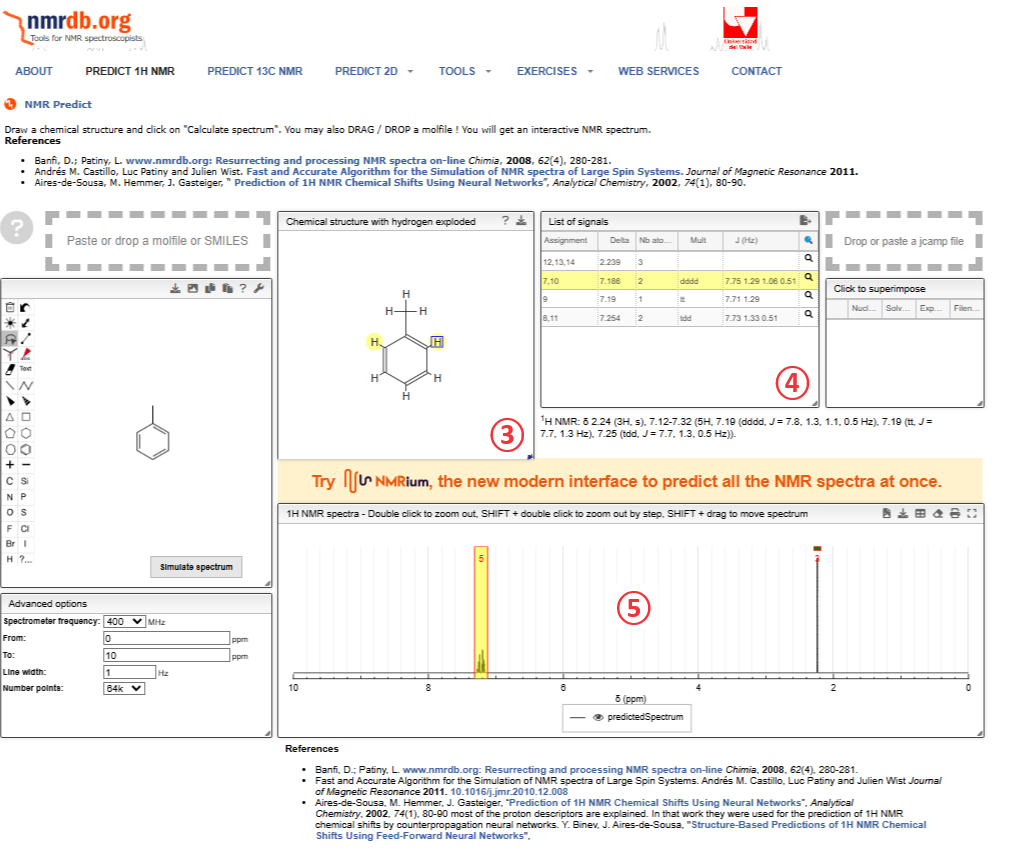
トルエンのNMR予想は,
メチル基が 2.239ppm
ベンゼン環の オルト位が 7.186 ppm
ベンゼン環の パラ位が 7.19 ppm
ベンゼン環の メタ位が 7.254 ppm
と予想される。
また,⑤のNMRチャートにおいて,ピーク位置でドラッグして範囲を決めるとピークを拡大できるし,NMRチャート内でダブルクリックすることでもとに戻すことができる。
さて,
SDBSの有機化学データベースで調べてみると,メチル基が「2.34ppm」でベンゼン環のプロトンはメチル基に対するプロトンの位置によらず「7.38ppm」となっている。
6.予想結果の活用の仕方
今回はトルエンを例にしたので,実際の測定結果と予想結果に大きな違いがあっても,背景の知識があれば概して利用できるが,
大事なことは
ベンゼン環に一つのメチル基(電子供与基)が付加した場合,オルト位>パラ位> メタ位 の順番でプロトンのピークが検出される可能性があるということである。
予想されたppmの位置は多少の誤差が出たとしても,「オルト位>パラ位> メタ位」の順番でプロトンのピークが検出される可能性が高いことを知れることが,実際の測定結果の解析に時短として使えるわけである。
例えば,
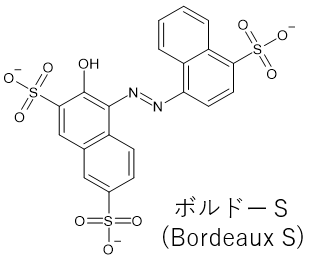
ボルドーSという色素分子がある。
先に紹介したYouTube動画を通して、ひとつずつプロトンを帰属しても良いが。
「分子の対称性を考慮して、こことここのプロトンは同じになるので。。。。」「大変!!」
このサイトを利用することで,一瞬でNMRスペクトルの予想をしてくれる。なんて、便利!
以下のような予想結果が出た。
この結果を基に,
どの順番でプロトンが並ぶか?!
がおおよその検討が付くので,実際のNMRスペクトルデータと照らし合わせることで解析のハードルが大幅に下がる。
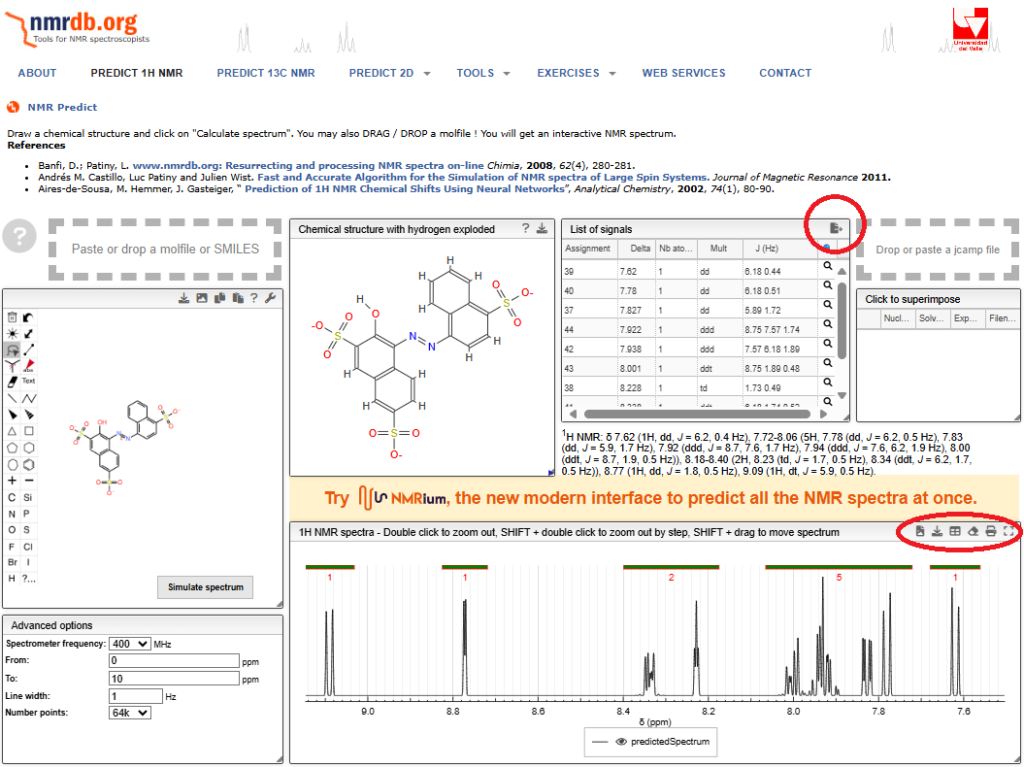
なお,上記の画像の「赤丸」からはテキストデータを得ることができるので,見ながらの手入力をする必要もないし,描画ソフトがあればNMRチャートを図としても活用できる。
7.もう一回使うとき
ウィンドウ①の左上に”ゴミ箱”アイコンがあるので、それをクリックすることで入力した構造式が削除されますので,新たに分子構造を入力できるようになります。
NMRの教材
自分勉強用に教材を紹介してくださいとありましたので,私が使っている教材をご紹介します。


応援!いつもありがとうございます。





